CH310M/318M Organic I
|
Dr. Brian
Pagenkopf |
|
|
|
Alkene Bromination
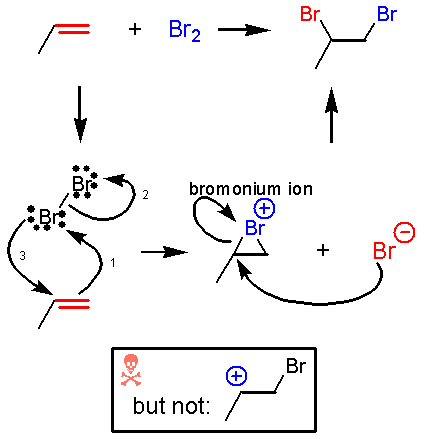
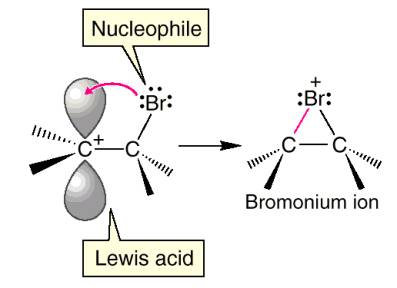
A lot happens in the first step of this reaction. The bromine-bromine bond is very weak. Attack by the alkene nucleophile (1) causes the bromine-bromine bond to break (2) so that bromine doesn’t end up with more than 8 valence electrons. Experiments suggest that there is no discrete carbocation intermediate, so the bromine must donate a pair of electrons to the other end of the alkene as it is being attacked (3). Instead of an intermediate cation, there is an intermediate bromonium ion that is then attacked by Br¯. The significance of an intermediate bromonium ion and not a carbocation is that the reaction is stereospecific.
Why
the bromonium ion? So why would bromine want to straddle two atoms and formally accept a
positive charge? The Golden Rules remind us that charge delocalization
is better than charges on a single atom, and a lot of partial positive charge
remains on the carbon atoms bromine is attached to\. The other reason for formation of a bromonium
ion is that bromine is huge compared to a hydrogen. If there’s a carbocation next to a bromine
atom its lone pairs of electrons are already getting close enough to interact
with the positive charge just because it is so big. Since the bromine atom is going to be
interacting with an adjacent positive charge anyway, limiting the intermediate
to a formal bromonium ion lowers the energy of the entire system because it
allows more efficient orbital overlap and charge distribution.
How
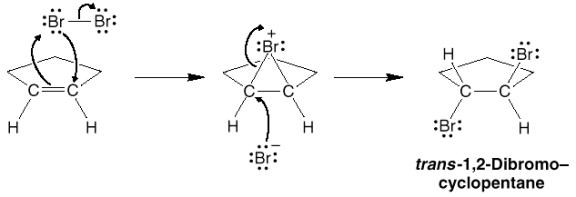
is this reaction stereospecific? If we look at
the bromination of methylcyclohexene, of the four possible products A through
D, only the enantiomers A and B are formed.
Note the stereochemical relationship of the bromines, they are anti
(or trans relative to each other). (Syn is analogous to cis in these cases).

The
selectivity observed in this reaction can be explained by looking closer at the
mechanism. Let’s use a simpler example,
bromination of cyclopentene.
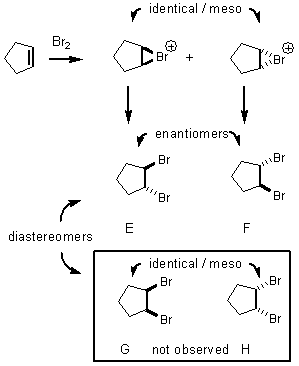
If
we guessed that an intermediate carbocation was formed, then all four
bromination products E through H would be expected. As we have already studied, nucleophiles can
attack the carbocation from either side of the p orbital.
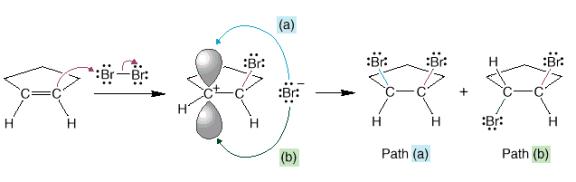
However,
in every case attack only happens from the opposite face from the bromonium
ion.
Why
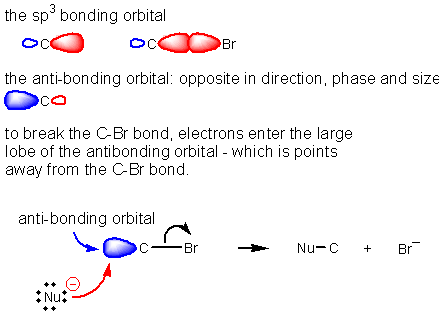
does the bromine always attack from the back side? Sterics alone isn’t enough to explain why the
nucleophile will come from the back side in every case, a molecular orbital
explanation can account for the perfect anti selectivity of this reaction. What orbital are the electrons from the Br¯
going into? Remember the electrons cant
go into a filled orbital, so they must go into the C-Br anti-bonding
orbital. The anti-bonding orbital points
away from the C-Br bond, and therefore incoming nucleophiles always attack from
the opposite or back side.
Mechanistically Similar Reactions:
Addition of ROBr (Br2 and H2O
or Alcohols)
Oxymercuration and reduction.
Addition
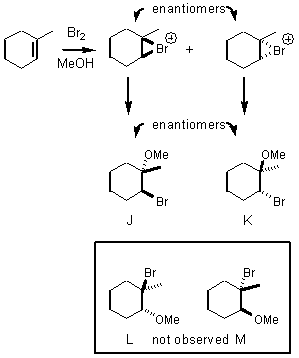
of HOBr. Let’s look at an example that’s similar to
the addition of Br2 to methylcyclohexene, except in this reaction
the solvent will be methanol. The
intermediate bromonium ion is intercepted by methanol to give the products J
and K. We expect an anti-relationship between the MeO and the Br because of the
bromonium ion intermediate, but note that none of the regioisomers L or M were
formed. It also appears that the
nucleophile MeOH went for the carbon with more steric hindrance!
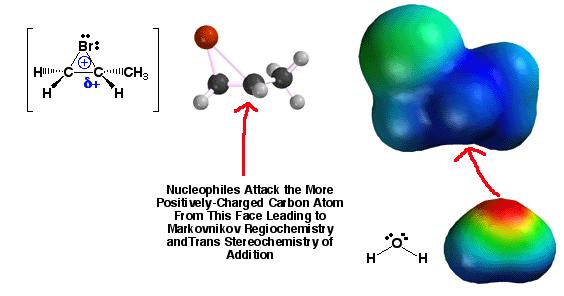
When
the two carbons of the halonium ion are not the same, more positive charge ends
up on the carbon attached to the greatest number of carbons. Or, the bromine ends up on the carbon with the
most hydrogens. That is, the addition of
HOBr follows Markovnikov’s rule. The
nucleophile (a base) attacks at the most Lewis acidic carbon, not the carbon
that is easiest to get to.