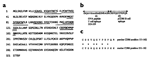Pig to human xenotransplantation provides a possible solution to the acute
and worsening shortage of donor organs. Enormous progress has been made in
recent years due to the creation of pigs transgenic for human complement
regulatory proteins. This single modification appears able to prevent hyperacute
rejection, and the survival of vascularized xenografts in nonhuman primates is
now measured in weeks rather than hours1-3.
However, there are several immunological barriers that must be overcome before
long-term survival of such grafts can be achieved. One obstacle is the strong T
cell immune response evoked by porcine tissues.
The T cell response to xenogeneic tissues involves two distinct pathways,
direct and indirect4, which have been well
documented for allorecognition and allograft rejection. The indirect pathway
refers to xenoantigens that are handled in exactly the same way as other protein
antigens. They are taken up, processed and presented by recipient antigen
presenting cells (APCs) in the form of peptides bound to recipient major
histocompatibility complex (MHC) molecules. This type of T cell response does
not involve any cross-species interactions in the generation of costimulatory
signals. The direct pathway results from CD4 and CD8 T cells interacting with
the xenogeneic MHC molecules in intact form on the surface of xenogeneic APC.
This will only culminate in recipient T cell activation if the relevant
interspecies molecular interactions occur with adequate efficiency to transduce
the appropriate signals. In vitro, both direct and indirect pathways of
xenorecognition induce strong primary proliferative responses by human T cells
against porcine stimulator cells4-11. Indeed, the
direct human anti-porcine T cell response is quantitatively comparable to direct
alloresponses between HLA-mismatched pairs, reflecting productive interactions
between key accessory molecules across the species barrier and the delivery of
costimulation by porcine B7 molecules through CD2812.
Although conventional immunosuppressive drugs appear to be able to prevent
cell-mediated rejection in preclinical models, the doses of drugs that are
likely required in the long term are unacceptable. Thus for the clinical
applicability of xenotransplantation, graft-specific strategies for tolerance or
immunosuppression would be highly advantageous. Despite all the problems of
discordant xenotransplantation, the use of organs from a disparate species does
create opportunities for donor-specific immunotherapy that allotransplantation
does not allow. This arises due to species-specific differences in key
immunostimulatory molecules.
One such molecule is CD86, a major costimulatory molecule in T cell
activation. The crucial role played by costimulatory molecules in determining
the result of T cell receptor (TCR)-CD3 receptor engagement with MHC and
peptides has been demonstrated extensively both in vitro and in vivo
in the context of both allotransplantation and xenotransplantation13-20.
For example, the use of a B7-binding fusion protein, CTLA4-Ig, to block
signaling via CD28-B7 resulted in enhanced graft survival and the prevention of
chronic rejection in a rat cardiac allograft model and a murine aortic allograft
model16-18. In these studies, administration of
CTLA4-Ig caused partial or complete tolerance to graft antigen. It has also been
demonstrated that treatment of allogeneic pancreatic islet transplant recipients
with antibodies to CD80 and CD86 inhibits transplant rejection13.
In the realm of xenotransplantation, Lenschow et al. have demonstrated
long-term donor-specific tolerance of human islets transplanted into mice with
concomitant treatment with CTLA4-Ig19.
Graft-specific tolerance was demonstrated to be a direct consequence of
inhibiting recognition of B7-expressing APC. Thus, anticostimulatory molecule
strategies aimed at either the receptors or their ligands can be used as
therapeutic strategies for altering the immune response.
Previously we and others have shown that pCD86 efficiently costimulates human
T cells through human CD28, and is therefore an attractive target for immune
intervention12. Constitutive expression of pCD86
by porcine vascular endothelial cells is an additional factor contributing to
the antigenicity of the graft12 and thus the
direct antiporcine T cell response is unlikely to diminish with time, as appears
to occur following allotransplantation21, 22.
In contrast the graft itself will continue to stimulate the direct pathway of T
cell activation indefinitely. Conventional approaches to inhibiting the delivery
of costimulation involve the injection of antibodies or fusion proteins designed
to block the relevant interactions13, 14,
18-20.
The strategy we tested here is designed to capitalize on species differences
and to generate an endogenous, donor-specific, costimulation-blocking antibody
response. If successful this approach would avoid the need for a series of
post-transplant injections of biological reagents. We have employed a chimeric
peptide strategy to generate antibodies to pCD86 without priming T cells against
porcine antigens.
Results
Design of chimeric peptides
Nine putative pCD86 B cell epitopes were designed on the basis of antigenicity
and hydrophilicity plots corresponding to sequences predicted to lie on the
exposed surface of pCD86 (Fig.
1a). Each peptide incorporated regions of nonidentity between porcine and
murine protein sequences. Mice were immunized with pools of peptides and the
antisera screened for reactivity with individual peptides by ELISA. One of the
peptides, OVA-pCD86, was selected based on the strength of evoked antibody
response. OVA-pCD86 peptide is a 29mer comprising a known T cell epitope,
ovalbumin residues 323–339 (OVA(323–339)) and 12 amino acids from pCD86 (Fig.
1b). The OVA-pCD86 peptide sequence is located in the membrane proximal
domain of the CD86 predicted structure. OVA(323–339) and the pCD86 sequence
alone (referred to as pCD86 peptide) were used as control peptides for the
separate analysis of T and B cell responses. The pCD86 component of OVA-pCD86
peptide shows 50% sequence identity with the murine homolog (Fig.
1c). Of the six amino acid substitutions between the two species, only two
are conservative; serine to threonine at position 152 and arginine to lysine at
position 162. The sera from mice immunized with the peptides that failed to
evoke a strong anti-peptide response served as internal negative control sera.
pCD86 recognized in a species-specific manner
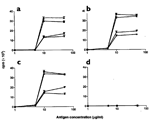
Two of the three mice injected with OVA-pCD86 peptide generated high titers of
specific antibodies to OVA-pCD86 peptide. Although one mouse responded with a
very low titer of OVA-pCD86 peptide antibody, all three mice generated OVA-pCD86
peptide antibody titers greater than those from OVA-immunized mice (Fig.
2a,b). Thus mice injected with OVA-pCD86 peptide are capable of generating a
specific antibody response to this peptide, but antibody titers differ between
the individual animals. These data are representative of many experiments
demonstrating specific antibody responses to OVA-pCD86 peptide. To confirm which
portion of the chimeric peptide was providing the antibody epitope, sera were
screened against pCD86 peptide alone. Sera from the OVA-pCD86 peptide mice
recognized the pCD86 peptide at a similar titer to the OVA-pCD86 peptide
containing both the T and B cell epitope (
Fig. 2c). This confirmed that the antibody epitope lay within the pCD86
sequence and did not involve the chimeric peptide junction.
To determine whether the antipeptide would recognize the native molecule from
which the peptide sequence was derived, sera from immunized mice were screened
against pCD86-expressing transfectants or the parent cell line. OVA-pCD86
peptide sera (1:25 dilution) clearly detected pCD86 on the cell surface (Fig.
3a) at a level comparable to that detected by 1 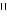 g/ml
of murine CTLA4-Ig (Fig.
3b). Sera failed to bind to the untransfected parent cell line (Fig.
3e). To further test the species-specificity of the sera, murine CD86
(mCD86) transfectants were screened using the same protocol. Although surface
expression of mCD86 was detected by murine CTLA4-Ig (Fig.
3d), OVA-pCD86 peptide antisera did not recognize native mCD86 (Fig.
1c), clearly demonstrating the species specificity of the sera. In all
circumstances, control OVA peptide antisera or human IgG1 gave negative results.
g/ml
of murine CTLA4-Ig (Fig.
3b). Sera failed to bind to the untransfected parent cell line (Fig.
3e). To further test the species-specificity of the sera, murine CD86
(mCD86) transfectants were screened using the same protocol. Although surface
expression of mCD86 was detected by murine CTLA4-Ig (Fig.
3d), OVA-pCD86 peptide antisera did not recognize native mCD86 (Fig.
1c), clearly demonstrating the species specificity of the sera. In all
circumstances, control OVA peptide antisera or human IgG1 gave negative results.
 |
High
resolution image and legend (47K) |
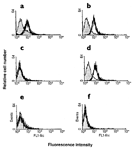
Figure 3.
Species-specific recognition of native pCD86 by OVA-pCD86 peptide
antisera.
Chinese hamster ovary (CHO) cells transfected with pCD86 (a,b)
or mCD86 (c,d), or the mock-transfected cells (e,f), were
stained with sera collected from OVA-pCD86 peptide–immunized mice
(thick lines in a,c,e) or OVA control peptide–immunized mice
(filled histograms in a,c,e). Antibody binding was determined by
two layer staining and assessed by flow cytometry. CTLA4-Ig staining
confirms expression of CD86 by both transfectants (thick lines in b,d)
compared to human IgG1 control (filled histograms in b,d), but
not on untransfected control cells (f).
|
|
T cell response is OVA(323–339)-specific
OVA-pCD86 peptide is a chimeric peptide containing a known T cell epitope and a
predicted B cell epitope derived from pCD86. For the success of our immunization
strategy it was imperative that mouse T cell responses were generated against
the ovalbumin sequence alone and not towards the native pCD86 molecule. T cell
responses directed against pCD86 would have been detrimental, accelerating graft
rejection.
To confirm T cell sensitization against the OVA peptide, and to exclude
sensitization against the pCD86 sequence, proliferation assays were done with T
cells from mice primed with ovalbumin and boosted with OVA-pCD86 peptide or a
variety of controls: OVA(323–339), pCD86 or ovalbumin alone. T cells were
rechallenged in vitro with the different antigens. Maximal T cell
proliferation was detected in mice boosted with either the OVA peptide or
OVA-pCD86 peptide when rechallenged with either ovalbumin, OVA peptide or
OVA-pCD86 peptide (Fig.
4a–c). In addition, mice primed with ovalbumin and boosted with pCD86
alone, failed to mount an antibody response in the absence of T cell help (data
not shown). Proliferative responses by T cells from pCD86 peptide-boosted mice
were no different from those of mice that received no boosting after the initial
priming with ovalbumin. Most importantly no proliferation was seen from any of
the mice in response to the pCD86 peptide. These data clearly demonstrate that T
cell responses in the immunized mice are directed towards the OVA epitope (
Fig. 4a–c).
Antisera specifically inhibit pCD86 costimulation
T cell proliferation assays were performed to determine whether OVA-pCD86
peptide antiserum was able to block CD86-mediated costimulation in vitro .
Purified CD4 T cells from DO.11.10 TCR transgenic mice were used as responder
cells. DO.11.10 T cells are specific for OVA(323–339) peptide in the context
of H-2Ad. CHO H-2Ad transfectants, supertransfected with
cDNAs encoding either pCD86 or mCD86, were used as the stimulator population.
Comparable levels of CD86 and H-2Ad were expressed on the two
transfected cell lines, as determined by flow cytometric analysis (data not
shown).
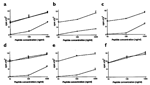
As illustrated in Fig.
5a, pCD86 appeared to costimulate primary responses by D0.11.10 T cells with
comparable efficiency to its murine counterpart. The proliferative responses to
both stimulator populations were inhibited to comparable extents by the presence
of either anti-MHC class II or murine CTLA4-Ig (Fig.
5a–d), further suggesting functional similarity between the two species of
CD86. However, the OVA-pCD86 peptide antisera had markedly different effects
when costimulation was provided by mCD86 or pCD86. At a 1:50 dilution the
antiserum was as efficient as CTLA4-Ig in inhibiting T cell proliferation
supported by pCD86. In contrast, the antiserum caused no inhibition when the APC
expressed mCD86, indicating precise species specificity.
Antibody to pCD86 prolongs pancreatic islet survival
Transplantation of porcine islets into C57BL/6 mice is a well established model
for studying T cell mediated xenograft rejection. The immunogenicity of the
porcine islets is enhanced by contamination of the islet clusters with passenger
leucocytes and endothelial cells that are CD86 positive23-25.
To determine whether antibodies generated by immunization with OVA-pCD86
peptide were capable of inhibiting costimulatory interactions in vivo, a
porcine islet transplant model was used. Mice that had previously undergone the
peptide immunization protocol were rendered diabetic by streptozotocin induction
on day 35. Four days later, 1000 pancreatic islets were transplanted under the
kidney capsule of all mice with a blood glucose reading of 20 mM/l or above.
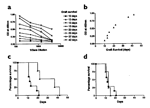
Our previous analysis of the antibody titers in OVA-pCD86 peptide–immunized
mice revealed mouse-to-mouse variation (Fig.
2a). We therefore measured the antibody levels in the sera of the
transplanted mice at the time of death. A range of antibody titers was detected
(
Fig. 6a) which correlated directly with graft survival time (Fig.
6b).
Having demonstrated a clear correlation between antibody titer and graft
survival time, the islet-grafted mice were divided into two groups, according to
the strength of their antibody response. High responders (OD >0.5 at sera
dilution 1:150, Fig.
6a, n = 4) and low responders (OD < 0.5 at sera dilution 1:150, Fig.
6a, n = 4). Survival curves for the high responders (Fig.
6c) compared to OVA peptide controls illustrate significant (P =
0.0121 as determined by Mann Whitney nonparametric statistical analysis)
prolongation of islet graft survival in OVA-pCD86 peptide–immunized mice,
median survival time 29 days (range 20–47 days) compared to OVA peptide
controls median survival time 12 days (range 11–21 days). In contrast,
survival curves for the low responders (Fig.
6d) compared to OVA controls show no significant difference.
Another experiment was performed to exclude the possibility that the higher
antibody titers in the mice were secondary to, rather than causative of, the
longer survival. A group of mice were immunized with a suboptimal concentration
of OVA-pCD86 peptide (50 g
instead of 100 g). Sera
were collected immediately before transplantation and again after mice were
killed following graft rejection. Suboptimal levels of OVA-pCD86 peptide failed
to prolong graft survival and antibody titers were of comparable levels pre- and
post-transplantation (data not shown). Peptide ELISA analysis of sera from
transplanted mice previously immunized with OVA peptide alone failed to
recognize peptide 6 in vitro , thus, the islet graft itself did not
induce or amplify anti–OVA-pCD86 peptide responses (data not shown).
Furthermore sera from post-transplant recipients screened by cell ELISA on
pCD86- or mCD86-transfectants only stained the pCD86-expressing cells,
confirming the continued specificity of our sera post-transplantation (data not
shown).
Discussion
The results presented here demonstrate that an endogenous costimulation-blocking
antibody response can be induced by peptide immunization, that this antibody
response was xenospecific, and that it led to the prolongation of survival of
porcine pancreatic islet xenografts in immunized mice. Parallel in vitro studies
demonstrated the ability of the peptide-induced antisera to inhibit direct
pathway mouse anti-pig T cell xenoresponses when costimulation was provided by
porcine, but not murine, CD86.
The significant prolongation of porcine islet graft survival in the immunized
mice was surprising for two reasons. First, the antibody response was only
generated against a sequence in pCD86, thereby leaving other costimulatory
molecules free to interact with murine ligands. No data exists concerning the
interaction between porcine CD80 and murine CD28 so it is possible that this
interaction is inefficient or nonexistent. The other obvious limitation of this
strategy is that it was only capable of inhibiting direct mouse anti-pig T cell
responses, triggered by recognition of intact porcine MHC molecules on the
surface of costimulation-positive porcine cells. Based on other species
combinations, including human anti-pig, the indirect mouse anti-pig T cell
xenoresponse is probably vigorous. Studies from our group have demonstrated both
direct and indirect responses of murine T cells against porcine stimulators in
vitro with comparable levels of responsiveness (data not shown). It is
likely therefore that islet graft failure in these experiments was mediated by
an unfettered indirect T cell response. In this study we have not examined the
fate of T cells with direct anti-pig specificity. It is attractive to imagine
that xenorecognition in the presence of costimulatory blockade may have induced
xenospecific nonresponsiveness, as has been observed in other experimental
settings.
Transplantation of nonvascularized tissues lends itself to the approach
described here, in that the parenchyma of the graft itself is not a target of
the induced antibodies. Whether or not the same approach could be used in
recipients of vascularized porcine xenografts is a matter of conjecture. It is
well established that porcine vascular endothelial cells expressed B7 family
molecules12. As a consequence, an induced anti-pB7
response would add to the burden of preformed antibodies to pig depositing on
porcine vascular endothelium immediately following transplantation. However,
based on the impressive results obtained with high level expression of
complement regulatory proteins, such as decay accelerating factor (DAF)1-3,
it may well be that the deleterious effects of antibodies to pB7 could be
adequately regulated in organs from DAF-transgenic pigs.
It can be further predicted that the antibody response will be sustained by
the graft for as long as the target antigen is expressed. This prediction was
fulfilled in another study aimed at achieving contraception by vaccination in
which mice were immunized with peptides incorporating sequences of mouse GNRH (gonadotrophin-releasing
hormone) and a T cell epitope. Peptide immunization succeeded in breaking
self-tolerance so that the mice made antibodies to GNRH. The antibody response
was sustained indefinitely, presumably by the presence of endogenous GNRH in the
recipient mice26. This is an anticipated benefit
of the use of this strategy in the context of xenotransplantation.
The approach described and tested here shows how the species differences that
are integral to xenotransplantation can be exploited for its benefit. Clearly
this approach could be extended to include other donor-specific molecules that
may contribute to recipient T cell activation. Obvious candidates include CD80
and CD40. The potential clinical utility of this strategy will require
experiments to be carried out in a preclinical model.
Methods
Antibodies and reagents. Unless otherwise stated, all reagents were
purchased from Sigma. The following fusion proteins and monoclonal antibodies
were used in T cell proliferation assays: murine CTLA4-Ig and human CTLA4-Ig
(both from R&D Systems), and anti–H-2Ad (M5/114) were used at
saturating concentrations of 10 g/ml.
Human IgG (The Binding Site, Birmingham, England) and rat IgG (Serotec, Raleigh,
NC) were used as controls. Biotinylated sheep anti-mouse IgG and
streptavidin-HRP (both from Zymed, San Diego) were used for peptide ELISA
studies and fluorescein isothiocyanate (FITC)-streptavidin (DAKO, Carpinteria,
CA) for flow cytometric analysis. Human CTLA4-Ig followed by anti-human IgG-biotin
and FITC-streptavidin was used to detect surface fluorescence of pCD86 and mCD86
on cell transfectants.
Chimeric peptides and OVA(323–339) were generated on a peptide synthesizer
(Genosys, Cambridge, England) and purified by HPLC to greater than 70% purity.
Lyophilized peptides were reconstituted in sterile water and used at 1 g/ml
diluted in PBS buffer for immunizations, and 10 g/ml
for coating wells in the peptide ELISA. OVA (albumin, chicken egg, Grade VII)
was resuspended in sterile water and diluted 1:1 in CFA for immunizations.
Generation of pCD86 cell transfectants. RNA was extracted from a
transformed porcine endothelial cell line, A8, using TRI-reagent (Sigma). mRNA
was then reverse transcribed and pCD86 amplified from the cDNA by 35 cycles of
PCR at 56 °C with 1.5 mM magnesium. The 5' and 3' primers
GCATGGATCCATGGGACTGAGTAACATTCTCTTTG and GCATGTCGACTTAAAAATCTGTAGTACTGTTGTC,
respectively, were designed on the basis of the published pCD86 sequence12
to overlie the start and stop codons. A 956-bp fragment was generated and
subcloned into the eukaryotic expression vector pci.neo (Invitrogen, Carlsbad,
CA). Stable CHO H-2Ad transfectants were generated using the standard
calcium phosphate precipitation27. Clones were
selected using dynabead purification and limiting dilution.
Cell culture. The CHO cell transfectants (CHO H-2A d
(ref. 28), CHO H-2AdmCD86 (ref. 28)
and CHO H-2AdpCD86) were maintained at 37 °C, 5% CO2 in
RPMI-1640 supplemented with 10% fetal calf serum, 100 g/ml
penicillin, 100 U/ml streptomycin and 2 mM L-glutamine. Medium was changed every
third day. Once confluent, cells were detached for subculture by treatment with
0.5% trypsin, 0.5% EDTA.
Peptide ELISA. This was carried out according to a method
described previously29. 96-well polystyrene
microtiter plates (Maxisorb, Nunc, Copenhagen) were coated with 50 g/ml
poly-l-lysine diluted in PBS buffer for 45 min at 37 °C, followed by 0.5% (v/v)
glutaraldehyde for 15 min at 37 °C. Plates were washed with PBS buffer, then 10
g/ml peptide was added
for 1 h at 37 °C. Unreacted aldehyde groups were then blocked by the addition
of 1 M glycine (pH 7.2) for 30 min at 37 °C. Nonspecific binding sites were
blocked with 2% bovine serum albumin (BSA) (w/v) in PBS buffer for 1 h at 37 °C.
The plates were washed three times with PBS buffer, serum was then added
(dilution range 1:150–4050) in PBS buffer containing 0.1% BSA for 1 h at 37 °C.
Biotinylated sheep anti-mouse IgG (1:8000) was then allowed to bind for 1 h at
37 °C, followed by the addition of HRP-conjugated streptavidin (1:4000) . The
plate was developed with TMB substrate (Cambridge Bioscience) and the reaction
stopped by the addition of 1 M H 2SO4. The absorbance was
measured at 450 nm using a microtiter plate reader. The ELISA wells were washed
extensively with PBS buffer containing 0.05% Tween-20 after each step.
Flow cytometric analysis. To examine the expression of surface
antigens, CHO cells transfected with pCD86, mCD86 or mock-transfectants (2.5
105) were incubated with 1:50 dilution of the appropriate sera for 30
min on ice. Bound sera was detected by incubation with sheep anti-mouse IgG-biotin
(1:500), 30 min on ice, followed by FITC-streptavidin, 30 min on ice. Cells were
washed twice with ice cold FACs buffer (PBS buffer, 1% fetal calf serum, 0.05%
sodium azide) after each step. Cells were fixed with 1 % paraformaldehyde in PBS
buffer and subsequently analyzed on a FACSCalibur flow cytometer (Becton
Dickinson, San Jose). Dead cells and debris were excluded by forward and side
scatter gating.
T cell proliferation assays. T cells were purified from the lymph
nodes and spleens of DO.11.10 transgenic mice or C57BL/6 mice by 50% lymphosep
density gradient centrifugation, 1250g for 20 min (Harlan, Sera-Lab,
Leicestershire, England) and complement mediated lysis using anti–MHC class II
(M5/114) antibody and rabbit complement (Cedarlane Laboratories, Hornby,
Canada). To establish the specific recognition of OVA(323–339) by T cells from
peptide-sensitized mice, 2
105 T cells and 2
105 APCs from C57BL/6 mice were then cultured for 3 days in the
presence of various antigens. The antigens were titrated from 0–50 g/ml.
To assess the effects of the anti-peptide sera on the delivery of costimulation,
2 104
DO.11.10 T cells were cultured for 48 h with CHO H-2Ad stimulator
cells transfected with either pCD86 or mCD86, in the presence of OVA(323-339)
peptide over a 10–1000 ng/ml range. Anti-peptide serum was added at a final
dilution of 1:50. T cell proliferation was measured after 48 h by the
incorporation of tritiated thymidine over a 16 h period. Data shown are
representative of at least three independent experiments with similar results.
In vivo immunization protocol. For peptide injections, 6-
to 8-week-old C57BL/6 mice were injected subcutaneously with whole OVA (50 g)
emulsified 1:1 in CFA. Mice then received 100 g
of OVA-pCD86 peptide or OVA(323–339) intravenously on days 14, 21 and 28. On
day 35 mice were either killed and their lymph nodes, spleen and blood
collected, or they were rendered diabetic by intraperitoneal injection of 250
mg/kg streptozotocin before transplantation of 1000 islets on day 39.
Isolation and transplantation of porcine pancreatic islets. Pancreatic
islets were obtained from adult female large white pigs weighing 200–250 kg.
Islets were isolated and purified according to published protocols30.
Islets were prepared by intraductal injection of collagenase solution (Type V, 2
mg/ml) and digestion at 37 °C. Islets were then purified by centrifugation of
the digested tissue over a discontinuous Euro-ficoll gradient. Purity of the
final preparation was confirmed by dithisone staining and was always greater
than 90%. Isolated islets were cultured overnight at 30 °C in 95% air and 5% CO2
in Medium199 supplemented as described. After overnight culture, 1000 handpicked
islets were transplanted under the kidney capsule of streptozotocin-treated
C57BL/6 mice. Animals were considered diabetic when their blood glucose was
greater than 20 mM/l. The function of transplanted islets was assessed by
biweekly measurements of blood glucose levels. Islets were considered rejected
following two consecutive blood glucose readings of 20 mM/l or above.
All in vivo experiments were performed in compliance with the relevant
laws and institutional guidelines.