URL: http://www.nature.com/cgi-taf/DynaPage.taf?file=/nature/journal/v409/n6822/full/409818a0_fs.html
Date accessed: 25 February 2001
Nature 409, 818 - 820 (2001) © Macmillan Publishers Ltd.
![]() 15
February 2001
15
February 2001
PEER BORK AND RICHARD COPLEY
Peer Bork
and Richard Copley are at EMBL, Meyerhofstrasse 1, 69012 Heidelberg, Germany.
e-mail: Richard.Copley@EMBL-Heidelberg.de
Two rough drafts of the human genome sequence are now published. Completion of the sequences lies ahead, but the implications for studying human diseases and for biotechnology are already profound.With the publication of the human genome sequence — described and analysed on page 860 of this issue1 and in this week's Science2 — we cross a border on the route to a better understanding of our biological selves. But unlike the previously published sequences of human chromosomes 21 and 22 (refs 3,4), the present sequences of the whole human genome are not considered complete. The bulk of the data make up what is called a 'rough draft'. So what is all the fuss about? What exactly does 'rough draft' mean, and what can we learn from sequences such as this?
In the draft from the publicly funded International Human Genome Sequencing Consortium1, around 90% of the gene-rich — euchromatic — portion of the genome has been sequenced and 'assembled', the term used to describe the process of using a computer to join up bits of sequence into a larger whole. Each base pair of this 90% was sequenced four times on average, ensuring reasonable precision. Only about a quarter of the whole genome is considered 'finished' — another bit of genomics jargon, which basically means that each base pair has been sequenced eight to ten times on average, with gaps in the sequence existing only because of the limitations of present technology. Nonetheless, the sequence of base pairs in the draft is very accurate, and is unlikely to change much; 91% of the euchromatin sequenced has an error rate of less than one base in 10,000 (ref. 1).
For the other draft, that produced by Celera Genomics2, a variety of methods suggest that between 88% and 93% of the euchromatin has been sequenced and assembled. But direct comparison of these numbers with the public consortium's draft is almost impossible — different procedures and measures were used to process the data and to estimate accuracy. Both projects also have sequence data that were not used in the assembly process, raising the real level of coverage by a few percentage points.
These numbers might seem rather arbitrary, but even when the first genome of an animal species was published5, it was clear that simple, practical finish lines do not exist (Box 1, Fig. 1). The present level of coverage of the human genome reflects the point where a shift of focus occurs, from sequencing the genome many times over to producing a high-quality, continuous sequence6. There is some way to go yet.
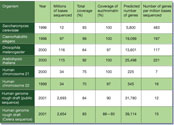 |
Figure 1
Sequenced eukaryotic genomes. Full legend High resolution image and legend (56k) |
Essentially, 'rough draft' refers to the fact that the sequences are not continuous — there are gaps (Box 1). If there are too many gaps, it can be impossible to order and orientate the many small strings of bases that are the raw products of genome sequencing. This might, for example, hamper projects that seek to identify genes involved in inherited diseases. A first step to finding such genes is to work out which region of which chromosome they are on. The complete genome sequence should be immensely useful for the next step — identifying the relevant gene at that region. But gaps and errors in ordering and placing the strings of sequence will make this difficult.
Another problem of incompleteness is that it is difficult to make definitive statements about which genes are unique to other species and do not have relatives in the human genome. So it might be prudent not to place too much emphasis on such 'missing' genes at this stage. Even so, they are running out of places to hide, particularly because the level of coverage of the human genome is probably higher than reported here1, 2 — there are other chunks of unassembled genome sequence in public databases, such as in independent collections of so-called expressed sequence tags.
But ensuring high quality and high coverage are only two aspects of producing a finished genome. For most biologists, the real interest is in the genes themselves. Here, the picture is less rosy, although the problems are caused not so much by the draft nature of the sequence as by the difficulty in finding genes among the other genomic DNA (Box 2).
Even coming up with a rough count of the number of genes is not straightforward. The public consortium's initial set contains about 32,000 genes, made up of around 15,000 known genes and 17,000 predictions. But these 32,000 genes are estimated to come from around 24,500 actual genes — some predicted genes could be 'pseudogenes', or just fragments of real genes. On the other hand, the sensitivity of prediction tends to be only about 60%, so it is reasonable to assume that another 6,800 or so genes (40% of 17,000) have been overlooked. This is how the present estimate of about 31,000 genes (6,800 plus 24,500) was reached1. Celera predicts that there are around 39,000 genes, but warns that the evidence for some 12,000 of these is weak2. The two groups use different gene-identification techniques, so these numbers are not directly comparable. Minor changes in procedures or data could alter either figure considerably. For example, such changes led to a recent estimate being lowered7, 8 from 120,000 to fewer than 81,000 — and both now seem untenable. Much is a matter of interpretation.
Fortunately, there is every reason to believe that the quality of gene prediction will rapidly improve, and an experimental technique for doing so is discussed on page 922 (ref. 9). With the sequencing of the genomes of other vertebrates, our ability to detect genes by their similarity to known sequences will get better. This is because, thanks to natural selection, gene sequences tend to be altered less during evolution than the DNA surrounding them. In a couple of years we should have at least a more complete list of testable gene candidates.
Despite all this, the information now available has profound implications. For example, there are already many heavily hunted disease-associated genes that have been identified using the public draft (ref. 1, Table 26, page 912). Together with studies of single nucleotide polymorphisms — the base differences from human to human — the draft also provides a framework for understanding the genetic basis and evolution of many human characteristics.
With the draft in hand, researchers have a new tool for studying the regulatory regions and networks of genes. Comparisons with other genomes should reveal common regulatory elements, and the environments of genes shared with other species may offer insight into function and regulation beyond the level of individual genes. The draft is also a starting point for studies of the three-dimensional packing of the genome into a cell's nucleus. Such packing is likely to influence gene regulation.
On a more applied note, the information can be used to exploit technologies such as chips made using DNA or proteins, complementing more traditional approaches. Such chips could now, for instance, contain all the members of a protein family, making it possible to find out which are active in particular diseased tissues. A new world of biotechnology will provide tools and information by exploiting genome data.
Sequencing the tough leftovers of the human genome will be essential. Without a finished sequence, we will not know what we are missing. Each missed gene is potentially a missed drug target, and even gene-poor areas might be critical for gene regulation. Nevertheless, we must now confront the fact that the era of rapid growth in human genomic information is over. The challenge we face is nothing less than understanding how this comparatively small set of genes creates the diversity of phenomena and characteristics that we see in human life. The human genome lies before us, ready for interpretation.
| 1. | International Human Genome Sequencing Consortium Nature 409, 860-921 (2001). |
| 2. | Venter, J. C. et al. Science 291, 1304-1351 (2001). |
| 3. | Dunham, I. et al. Nature 402, 489-495 (1999). | Article | PubMed | |
| 4. | The Chromosome 21 Mapping and Sequencing Consortium Nature 405, 311-319 (2000). |
| 5. | The C. elegans Sequencing Consortium Science 282, 2012-2018 (1998). |
| 6. | Collins, F. S. et al. Science 282, 682-689 (1998). | PubMed | |
| 7. | Liang, F. et al. Nature Genet. 25, 239-240 (2000). | Article | PubMed | |
| 8. | Liang, F. et al. Nature Genet. 26, 501 (2000). | Article | PubMed | |
| 9. | Shoemaker, D. D. et al. Nature 409, 922-927 (2001). | Article | |
| 10. | The Arabidopsis Sequencing Consortium Cell 100, 377-386 (2000). |
| 11. | Adams, M. D. et al. Science 287, 2185-2195 (2000). | Article | PubMed | |
| 12. | Normile, D. & Pennisi, E. Science 285, 2038-2039 (1999). | Article | PubMed | |
| 13. | Aparicio, S. Nature Genet. 25, 129-130 (2000). | Article | PubMed | |
| 14. | Goffeau, A. et al. Nature 387 (suppl.), 1-105 (1997). |
| 15. | The Arabidopsis Genome Initiative Nature 408, 796-815 (2000). |
Category: 32. Genome Project and Genomics